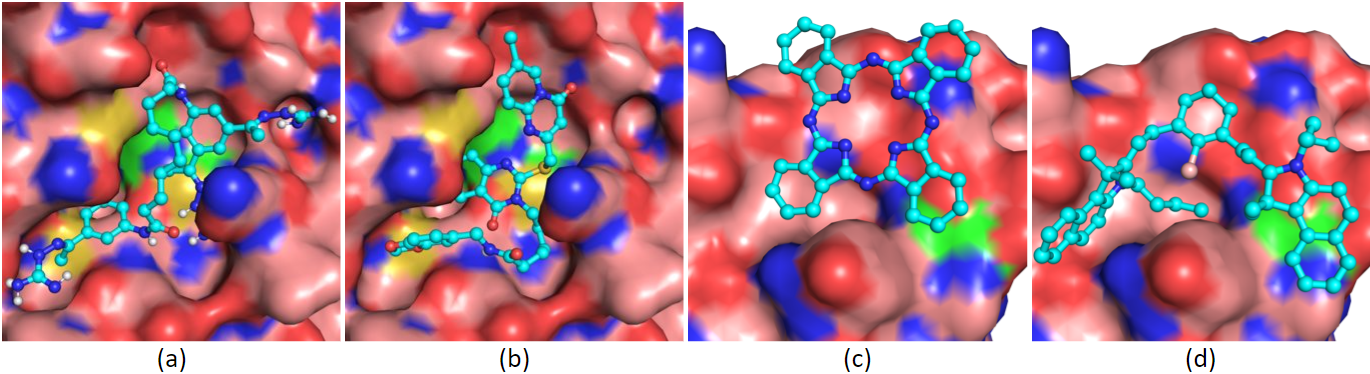
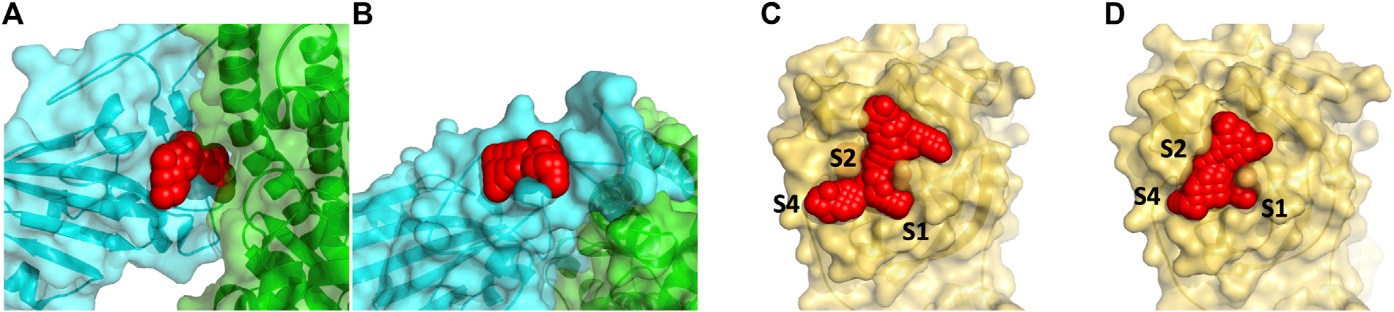
Initially intended for designing effective cancer drugs, ATOM’s modeling pipeline screens candidate drugs for predicted binding to target proteins. The high-throughput process was applied to COVID-19, using “docking” software to try to fit 1.6 billion small molecules into each of four SARS-CoV-2 protein “pockets.”
Given the almost 60 billion possible orientations of molecules within protein sites, the pipeline uses ML models to score each molecule based on multiple predicted properties: efficacy in killing the virus, possible toxic side effects, how well it is absorbed and distributed in the body, and how easily it can be synthesized in a lab. Scoring helps prioritize which molecules to pursue further.
“Traditional drug discovery involves many time-consuming and expensive experimental steps,” states computational biologist Kevin McLoughlin. Experimentalists choose a biological target such as a protein; test chemical libraries with screening assays to identify good candidates; design and synthesize variants of the top compounds; and test for potency, safety, and other properties. Virtual screening takes the place of physical, brute-force screening and opens up an extremely large design space by analyzing a vast number of possible candidates against their targets.
Thus, this virtual screening approach saves experimentalists time by radically narrowing the list of promising molecules that may inhibit specific viral protein targets. Faster time to experimentation means faster development of antiviral drugs that could treat or prevent COVID-19 infections. The team’s screening techniques have been published in Frontiers in Molecular Biosciences and accepted to SC21.
According to Lightstone, the team is aiming for a 7% success rate—10 times greater than the rate of physical screening of far fewer (tens of thousands) compounds by experimental groups around the globe.
Allen notes, “We’re in a much better position to propose new molecules that have potential to be valuable, but synthesizing them is a real practical challenge. More accurate prediction algorithms and more effective searching of the chemical space will be key.”